Introduction to the world of digital PCR
May 22, 2024 | Informative Articles

Featured in this article:
• Basics of PCR
• Why is it called digital PCR?
• How does dPCR work?
• Areas of applications of dPCR
• Summary of advantages of dPCR
Digital polymerase chain reaction (dPCR) is an extension of the conventional PCR method, which is used for the quantification and monoclonal amplification of nucleic acids. In other words, the amplification of nucleic acids that can be traced back to a common nucleic acid source. Nucleic acids include DNA, cDNA, methylated DNA and RNA. The main difference between dPCR and conventional PCR lies in the method of measuring the amount of nucleic acid. In conventional PCR, one reaction is carried out per prepared sample, whereas dPCR is able to divide the target nucleic acid molecules of the sample evenly into numerous parallel individual PCR reactions and amplify them. The separation of the target nucleic acid molecules results in absolute quantification. This means that the separation enables sensitive measurement and reliable determination of the amount of nucleic acid [1].
Basics of PCR
In general, the PCR method is used to amplify nucleic acid molecules. Various components are required for this, the DNA polymerase, the enzyme that catalyses the linking of nucleotides to synthesise nucleic acids, and the master mix, which usually consists of primers, nucleotides (dNTPs), cofactor (usally magnesium chloride; MgCl2), buffers and PCR grade water.
During the amplification of nucleic acids, an exponential increase of the target DNA molecules takes place within the PCR cycles in the classical sense.
This means that the initial quantity of nucleic acid is multiplied by a certain factor per PCR cycle.
lightbulb The first amplification cycles of a PCR may not be exponential [1].
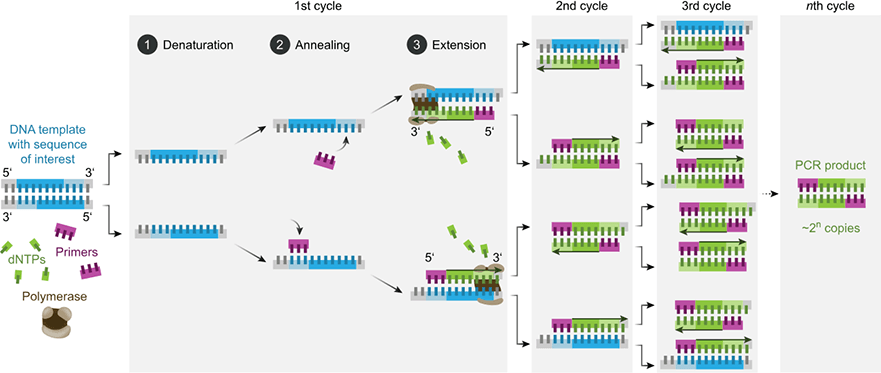 Image: WiWiki, CC BY-SA 4.0, via Wikimedia Commons | Enzoklop
Image: WiWiki, CC BY-SA 4.0, via Wikimedia Commons | Enzoklop
Figure: Fundamentals of PCR. Each PCR cycle consists of three steps: The denaturation of the double-stranded DNA by heating, the annealing of the primers to their complementary target DNA sequences, and the extension of the primers by a thermostable DNA polymerase. A PCR reaction consists of 20 to 45 cycles. Theoretically, each cycle can lead to a doubling of the number of molecules in the target sequence [2].
The starting DNA is initially present as double-stranded DNA, with the direction of the arrow pointing from 5‘ to 3’. After denaturation, the individual strands are separated so that the primers can bind. The polymerase extends the primers in the direction 5‘ to 3’, creating the opposite strand. The resulting products are still too long at one end, as only the starting point (primer) is defined, but not the end. In the next cycle, PCR products of the correct length are produced for the first time, although the counter-strands are still too long. Only in the third cycle is the PCR product formed as a double strand of the correct length. In the following cycles, the desired products multiply exponentially, as they serve as a template for further syntheses. In contrast, the unwanted long products from the first cycle only increase linearly, as only the DNA used serves as a template [2].
The exact determination of the initial DNA quantity of a sample cannot be carried out with conventional PCR. The informative value of conventional PCR is limited to answering the question of whether target DNA molecules are present or not. There are many factors that lead to uncertainties and inaccuracies in the exact calculation of the amount of starting material in a sample. PCR amplification comes to a standstill after an indefinite number of cycles. By-products and inhibitors can impair the activity of the polymerase or interfere with the binding of the primers to the target sequence. In addition, there may be variations in PCR amplification efficiency in different biological samples such as environmental blood and tissue samples. Biological samples may contain different initial concentrations of the target DNA molecules or inhibitory impurities [1].
lightbulb A low initial concentration of target DNA molecules can lead to the fact that a detectable level cannot be reached during amplification.
Why is it called digital PCR?

Figure: 24-well nanoplate
In this case, digital stands for the separation of the PCR reaction mixture (nucleic acid, master mix, assay reagents) into partitions. This means that the nucleic acids are divided into small compartments, resulting in sub-reactions [3]. The PCR reaction mixtures are loaded into the wells. The result is a thousand individual PCR reactions that are amplified on nanofluidic chips of the dPCR nanoplate or other partitioning methods (e.g. digital droplet PCR; ddPCR) and then classified as positive or negative reactions, which forms the basis for a digital output format [6]. Partitioning overcomes the limitations of conventional PCR [4].
lightbulb Partition is the sub-reaction used for limiting dilution and subsequently measured as a positive or negative signal. Each partition acts as an individual PCR microreactor and partitions containing amplified target sequences are detected by fluorescence [3].
How does dPCR work?
The target nucleic acids in the sample to be analysed are present as individual molecules in the partitions due to dilution during sample preparation. An individual PCR reaction is carried out in each partition, which is then analysed quantitatively. By partitioning the target nucleic acid, the number of target nucleic acids can be determined by estimation according to the Poisson distribution.
lightbulb Poissons distribution is the statistical distribution used to account for the probability that a partition initially contains more than one target nucleic acid [3].
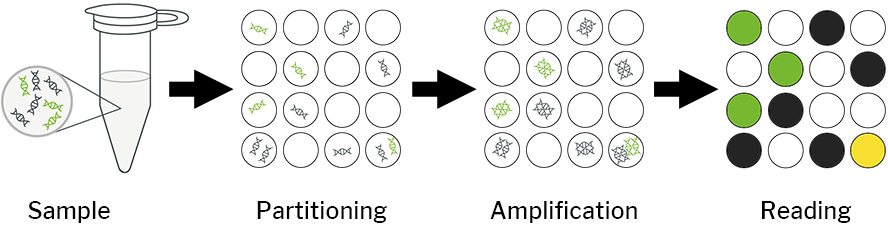
Partitions containing at least one nucleic acid target generate a positive signal, whereas partitions containing no nucleic acids generate a negative result [4]. This is based on the assumption that the number of molecules follows a Poisson distribution, taking into account that there may be several targets in a partition. The quantification of the amount of nucleic acid is obtained by counting the positive reactions after amplification of the samples in the final PCR product. In conventional PCR, the initial copy number is proportional to the number of PCR amplification cycles. In contrast, dPCR does not require the number of amplification cycles to determine the initial sample quantity. This eliminates the dependence on uncertain and inaccurate exponential data for quantification of target nucleic acids and enables absolute quantification [1].
lightbulb Dilution to very small amounts per reaction improves the proportion of rare species in a given reaction volume, even if the average proportion remains the same [6].
Areas of applications of dPCR
• Absolute quantification of viral load
• Absolute quantification of nucleic acid standards
• Absolute quantification of next-gen sequencing libraries
• Rare allele detection
• Low-fold copy number discrimination
• Enrichment and separation of mixtures [1]

lightbulb The dPCR is particularly suitable for applications that require both high validity and precise determinations.
Summary of advantages of dPCR
• No dependence on references or standards
• Increasing the total number of PCR repeats enables the desired precision
• Very robust method that is highly tolerant to PCR inhibitors
• Complex mixtures can be analysed
• dPCR provides a linear response to the number of copies present, allowing even the smallest differences in fold change to be recognised [1]
The dPCR simplifies the quantification of a target sequence by counting positive and negative amplification reactions and converting these signals into binary or digital formats. Advances in microfluidics have advanced this technology by providing effective methods for generating many independent reactors [5].
The process is increasingly being applied to projects that require high sensitivity and will quickly become the standard method. As technology continues to advance, the devices will continue to improve in terms of ease of use, higher partition density and larger reaction volumes. This will reduce costs and allow more laboratories access to this technology [6].
Stay tuned and discover more articles about molecular biological detection methods and their applications in research, diagnostics and industry!
Be the first to know and subscribe to our newsletter below!
[1] Pfaffl, Michael W. (n.d.): digital PCR on www.Gene-Quantification.Info, [online] https://www.gene-quantification.de/digital-index.html. [2] Wikipedia: Polymerase chain reaction (https://en.wikipedia.org/wiki/Polymerase_chain_reaction) [3] Whale, Alexandra S et al. (2020): The Digital MIQE Guidelines Update: Minimum information for publication of Quantitative Digital PCR experiments for 2020, in: Clinical Chemistry, vol. 66, no. 8, pp. 1012–1029, [online] doi:10.1093/clinchem/hvaa125. [4] Arnemann, J. (2019): Digitale PCR, in: Springer Reference Medizin, p. 700, [online] doi:10.1007/978-3-662-48986-4_3461. [5] Quan, Phenix-Lan et al. (2018): DPCR: a Technology Review, in: Sensors, vol. 18, no. 4, p. 1271, [online] doi:10.3390/s18041271. [6] dPCR – the digital polymerase chain reaction (2017): in: Analytical Methods, vol. 9, no. 29, pp. 4225–4227, [online] doi:10.1039/c7ay90093g.